Veamos hoy las simulaciones condicionadas a genotipos, que
se pueden hacer desde el módulo DVI de Familias. Como os anuncié en el post del
11 de noviembre (Simulando perfiles genéticos) podemos hacer este tipo de
simulaciones gracias a una herramienta incluida en Familias que se llama
Paramlink, y que fue desarrollada por el inquieto Magnus Vigeland (un
matemático del grupo de Thore que transmite mucha alegría  ).
).
Veo muy útil este tipo de simulaciones para los casos de
DVI, pero sobre todo para la identificación de personas tras conflictos armados
del pasado, pues en estos casos, los familiares de las personas desparecidas que
están disponibles en la actualidad no suelen ser familiares cercanos. Y no
sabemos si dispondremos de información genética de referencia suficiente para
lograr una identificación.
Con esta herramienta podréis saber si un pedigrí concreto
que ya tenéis analizado es suficientemente informativo o no, es decir, os
ayudará a saber a priori si vais a obtener un buen LR en el caso de que el
perfil de la persona desaparecida se encuentre en vuestra base de datos de
víctimas. La diferencia entonces entre este tipo de simulaciones y las simulaciones
no condicionadas a genotipos es fácil de entender: en las primeras sólo
evaluamos hipótesis en general y en las segundas además de las hipótesis vamos
a evaluar los perfiles genéticos de un pedigrí en concreto.
Paramlink ya se ha utilizado en el caso de las
desapariciones de Argentina, en el cual las abuelas están buscando a sus
nietos. Podéis verlo en este interesantísimo paper publicado recientemente: Kilng
D., Egeland T., Herrera M., Vigeland MD. (2017) Evaluating the
statistical power of DNA-based identification, exemplified by ‘The missing
grandchildren of Argentina’. FSI:Gen 31,
57-66.
Este ejemplo que os pongo es precisamente de este paper.
Imaginaros que tenemos muestras de los siguientes familiares de una mujer a la
que estamos buscando: un tío paterno, la abuela paterna, una hermana del abuelo
paterno y un bisabuelo paterno. Hemos analizado 15 marcadores. Para verlo más
claro:
 Los individuos sombreados, son los que hemos genotipado. MP
Los individuos sombreados, son los que hemos genotipado. MP
significa “missing person” y POI significa “person of interest”. Con Paramlink,
podemos simular:
a) perfiles genéticos que “encajen” en esta familia (H1 en
la figura) y
b) perfiles genéticos que “no encajen” en este pedigrí (H2
en la figura)
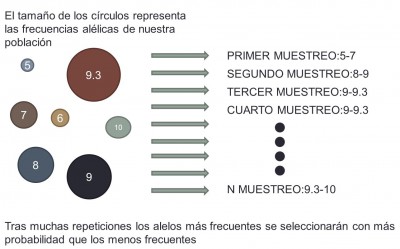
Como siempre, los perfiles simulados se generarán teniendo
en cuenta las frecuencias alélicas de nuestra población. En este caso entonces
sólo se simula el perfil de la persona desaparecida (encaje o no es esta
familia), los perfiles de los familiares NO se simulan, son los que nos
resultaron al analizar las muestras. Y Paramlink nos calculará los LRs. En el
paper nos ofrecen una gráfica con los resultados:

Nota: los valores de LR están en escala logarítmica, es
decir, 10 elevado al valor que aparece en el eje X. El eje Y representa la
frecuencia con la que aparece cada valor de LR, es lo que los matemáticos
llaman función de densidad y tienen la costumbre de no poner números en este
eje; es sólo para que veamos qué valores de LR son más frecuentes y cuáles son
menos frecuentes (cosas de matemáticos, esto de no poner números en el eje Y…parece
ser que los valores son obvios… será para ellos, porque para mí son una
incógnita,  ).
).
Bueno, a lo que nos interesa, ¿Qué significa esta gráfica?
Pues sobre todo nos dice dos cosas importantes:
a) Que es muy poco probable que identifiquemos mal, que
digamos que una mujer concreta es la persona desaparecida sin serlo realmente (falso
positivo). Es la intersección de las dos curvas que veis en la gráfica, y los
valores de LR en esa intersección van de 10^-2 (0.01) a 10^+2 (100), más o
menos. Y esos LRs no nos convencen…
b) Que si una mujer encaja en el pedigrí porque realmente sea la
persona desaparecida, vamos a obtener LRs mayores a 10^+4 (10.000) en muy pocos
casos, o lo que es lo mismo, vamos a necesitar más familiares y/o tendremos que
analizar más marcadores. Lo sabemos porque en la curva de línea contínua
(MP=POI) llegamos al 4 del eje X con poca frecuencia (pocas veces, tras todas
las simulaciones que hemos hecho).
Yo aún no he probado esta herramienta, la experta hispano-parlante
en esto es Mariana Herrera, así que ella os podrá contar mejor que yo!!
Enhorabuena Mariana por este fantástico paper!! Y como siempre, el eterno
agradecimiento a Magnus, Daniel y Thore!!